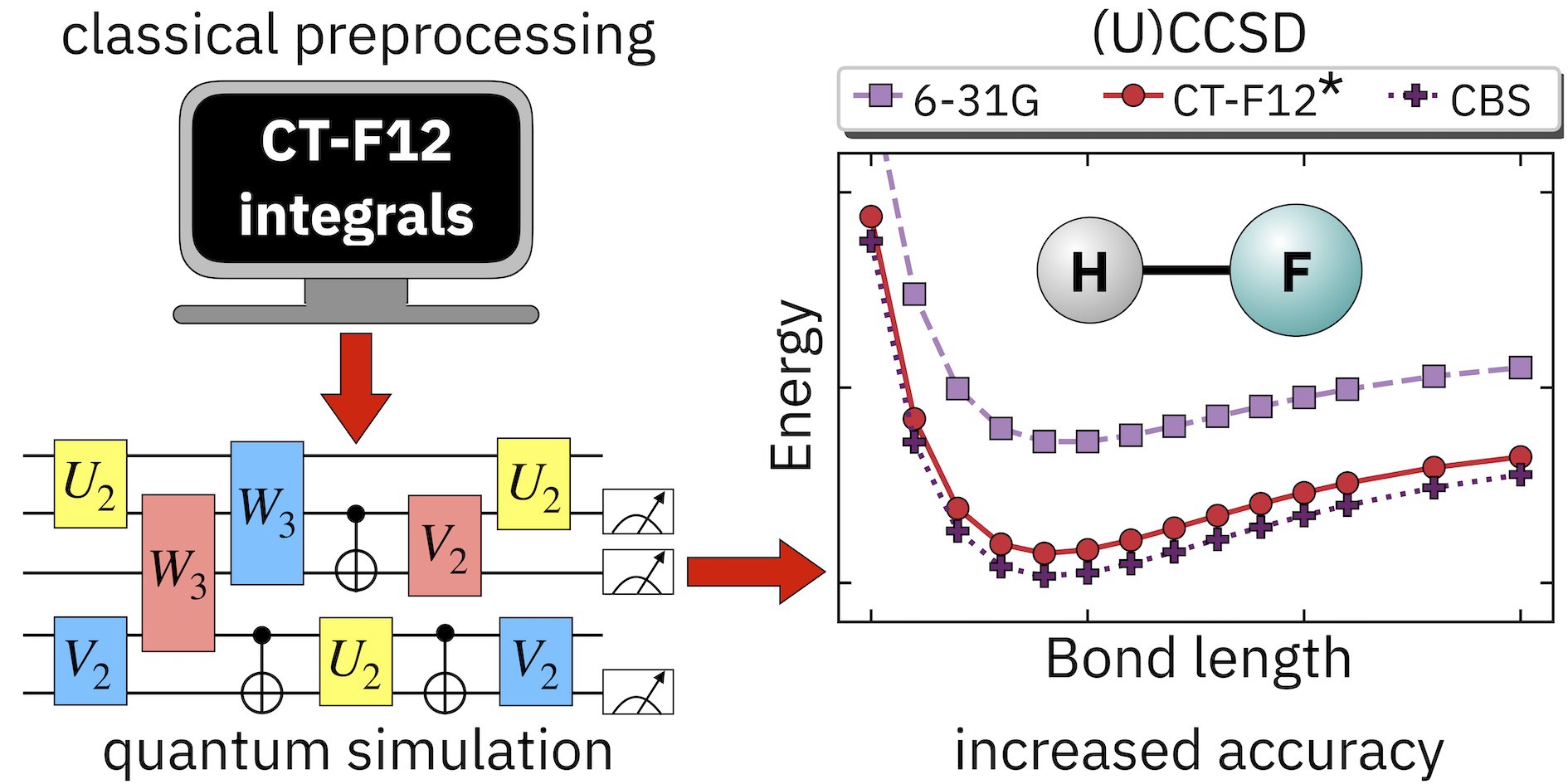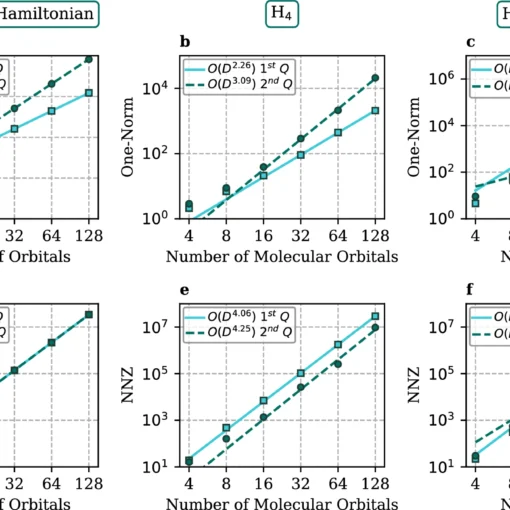
First-Quantization: A Quantum Leap for Molecular Energy Solvers
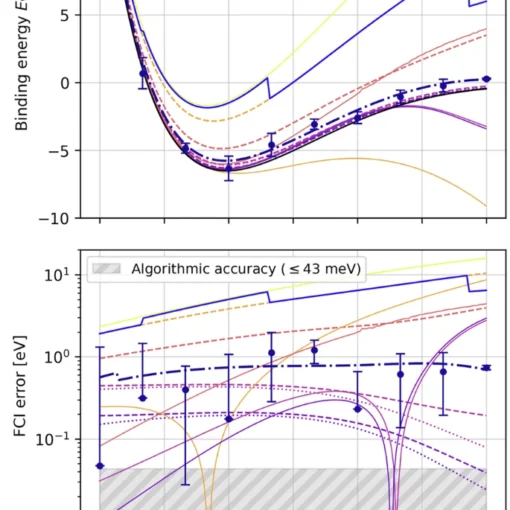
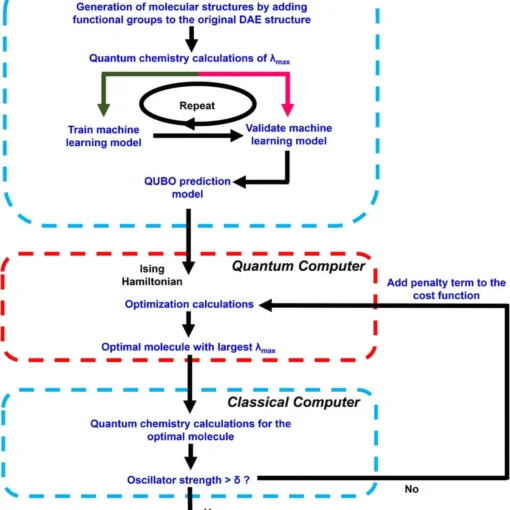
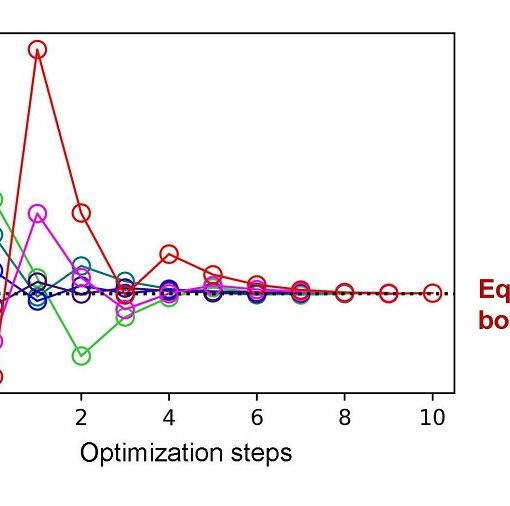
Researchers have developed a novel first-quantization method for quantum computing that achieves significant improvements in efficiency for molecular energy calculations across arbitrary basis sets, demonstrating both asymptotic speedup in molecular orbital calculations and orders-of-magnitude resource reductions when using dual plane waves compared to second-quantization approaches.