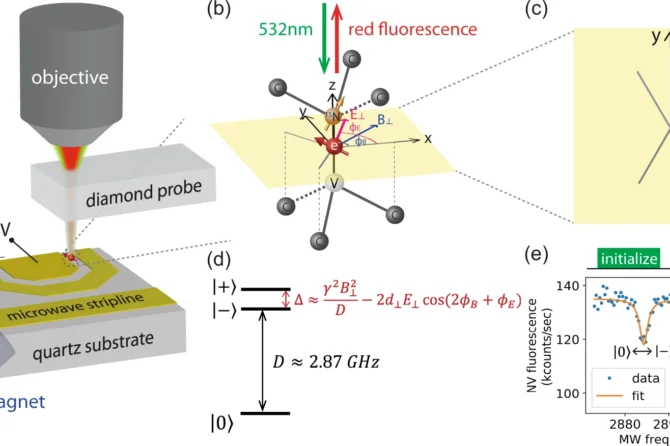
Quantum simulation of quantum chemistry is one of the most compelling applications of quantum computing. It is of particular importance in areas ranging from materials science, biochemistry, and condensed matter physics.
A team of researchers proposes a full quantum eigensolver (FQE) algorithm to calculate the molecular ground energies and electronic structures using quantum gradient descent.
Compared to existing classical-quantum hybrid methods such as variational quantum eigensolver (VQE), this method removes the classical optimizer and performs all the calculations on a quantum computer with faster convergence.
The gradient descent iteration depth has a favorable complexity that is logarithmically dependent on the system size and inverse of the precision. Moreover, the FQE can be further simplified by exploiting a perturbation theory for the calculations of intermediate matrix elements and obtaining results with a precision that satisfies the requirement of chemistry application.
The full quantum eigensolver can be implemented on a near-term quantum computer. With the rapid development of quantum computing hardware, the FQE provides an efficient and powerful tool to solve quantum chemistry problems.
Read more.