As the search continues for useful applications of noisy intermediate scale quantum devices, variational simulations of fermionic systems remain one of the most promising directions.
A team at Google AI Quantum has performed a series of quantum simulations of chemistry which involve twice the number of qubits and more than ten times the number of gates as the largest prior experiments.
The researchers used a variational quantum eigensolver (VQE) simulation with a Google Sycamore quantum processor.
They have modeled the binding energy of H6, H8, H10 and H12 chains as well as the isomerization of diazene. They also demonstrated error-mitigation strategies based on N-representability which dramatically improve the effective fidelity of our experiments.
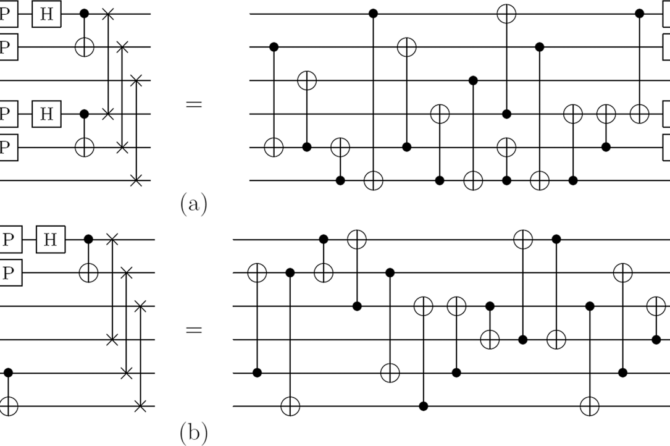
Their parameterized ansatz circuits realized the Givens rotation approach to free fermion evolution, which they variationally optimized to prepare the Hartree-Fock wave-function. This ubiquitous algorithmic primitive corresponds to a rotation of the orbital basis and is required by many proposals for correlated simulations of molecules and Hubbard models.
Because free fermion evolutions are classically tractable to simulate, yet still generate highly entangled states over the computational basis, they used these experiments to benchmark the performance of their hardware while establishing a foundation for scaling up more complex correlated quantum simulations of chemistry.